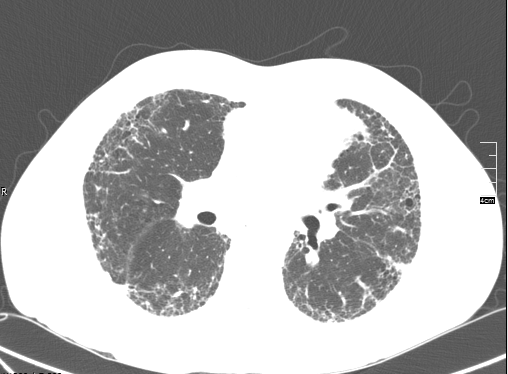
Pulmonary fibrosis is a somewhat generic term that describes scarring in the lungs. Idiopathic pulmonary fibrosis (IPF) is a very specific term that describes a specific disease process that leads to progressive scarring in the lungs. During the evaluation of scarring in the lungs, your doctor considers a variety of diseases that can cause scarring in the lungs. Two of the most common are IPF and non-specific interstitial pneumonitis (NSIP). Both diseases cause cough and shortness of breath. Both diseases lead to abnormal CT scans of the lungs. However, there are some very important differences.